Gibbs Free Energy - Video Tutorials & Practice Problems
On a tight schedule?
Get a 10 bullets summary of the topic
Time for Gibbs Free Energy, the most important equation for understanding reaction favorability! It’s going to be important that we understand the significance of all the terms in this equation.
1
concept
Breaking down the different terms of the Gibbs Free Energy equation.
Video duration:
5m
Play a video:
we know that the thermal dynamics or spontaneity of reaction is directly related to the Gibbs Free Energy. So it's gonna be really important that we understand how to break down the Gibbs free energy equation and know how to use it. All right, so in this video, what I'm gonna do is I'm basically just gonna describe every term and help you guys see how it relates to reactions. Okay, so the first thing just going all the way back gives free energy, or Delta G is going to predict the favorability of the reaction. Okay, remember that another word for favorability is spontaneity. Alright? And it's comprised of three terms. It's comprised of the Delta H, the Delta S and the T. So what I want to go through is just each 11 at a time and talk about what they mean. All right, so the delta H is the anthem API. Okay. Entropy can get confusing because Delta H and Delta G. A lot of times they happen to be the same. A lot of times, if you have a negative entropy, you also have a negative spontaneity. And a lot of students get confused thinking that they're actually the same thing. They're not okay. The Delta h is just one component off the spontaneity. But we also have to take into account the temperature and the delta s or the T in the delta s. So let me just start right there. Okay? What is the entropy? It's simply the some off the bond association energies for the reaction. So what that means is I'm gonna be making bonds. I'm gonna be breaking bonds. All those actions require that I'm putting in energy or I'm receiving some energy. Alright. When I add all those together, whatever my end number is, that's my entropy. Alright, that's it. That's all it has to do with. All right, So, um, later on, we're gonna learn how to actually add and subtract that stuff. But for right now, I'm just trying to tell you guys the big picture. Okay, So if something has a negative entropy, a negative delta H, that's what happens when you make bonds. Okay. When you make bonds, you are getting some energy back. Why? Because I already taught you guys. Remember that I showed you guys in the free energy free free energy diagram how you could gain energy by putting two atoms together. Okay, that's what we call exo. Thermic. Okay, exo. Thermic doesn't always mean that the reaction is favorite. Remember, favorability has to do with eggs, organic or inorganic, but it is one component of it. Okay, well, what if your anthem Pete is positive? If it's positive, that means I'm breaking bonds because it requires energy to break bonds. Okay, once you break bond, that's gonna be endo thermic because of the fact that it requires that in putting energy into the system to make those atoms separate. Okay, that's all the entropy is. It's just the some of these the endo thermic parts and the exo thermic parts together. And then at the end, you see if the overall number is negative or positive, and that determines your adult age, it's not to talk about one. That's actually a little bit more difficult to understand. Is the entropy, the Delta s the men? The entropy is a measure of disorder in the system, all right, And this consume very confusing because it's like, how does the system know how disorder it is? I'm gonna explain this in more depth later when I actually talk about in tribute on all in its own. But for right now, I just want you guys to know what the signs mean. And if it's negative entropy, that means that I'm going to, um, or ordered state. Okay, that means that I'm basically entropy means how disorder something is. So it's the opposite of disorder that's ordered. If it's positive, that means it's more disordered. Okay, a state always wants to be in its most disordered possible arrangement possible. So it's gonna be a good thing. Okay, So when Delta s is positive, that means my reaction is gonna be a little bit more favored. Alright. So hopefully guys understand that negative means ordered positive means disordered. Positive is the one that is actually favored. Okay, then finally have her last variable, which is temperature and notice where temperature is in the equation. Temperature is gonna be a coefficient of delta s. And what that means is that as temperature goes up, it's going to amplify the effect of entropy on my overall favorability. What that means is that as I jack up the temperature of my reaction, the entropy of the reaction is gonna is gonna matter a whole lot more in determining the overall, um, fate of my reaction, whether it's gonna be favorite or not. Okay. On the other hand, as I reduce my temperature down toe zero Kelvin or absolute zero, it's gonna mean that my entropy becomes less and less important. Okay, Eventually, once you, as you approach absolute zero entropy doesn't matter at all anymore because there's actually no more movement. And all that matters is the heat of dissociation. Okay, that's kind of theoretical, but I hope that makes sense to you guys. We're gonna be learning and talking about that more later when we discuss entropy all on its own.
1. Enthalpy (ΔH°) is the sum of bond dissociation energies for the reaction.
Negative values (-) indicate the making of new bonds = Exothermic
Positive values (+) indicate the breaking of new bonds = Endothermic
2. Entropy (ΔS) is the tendency of a system to take its most probable form.
Negative values (-) indicate less probable = Unfavored
Positive values (+) indicate more probable = Favored
3. Temperature (T) amplifies the effect of entropy on the overall favorability.
2
concept
Intermediates vs. Transition States
Video duration:
6m
Play a video:
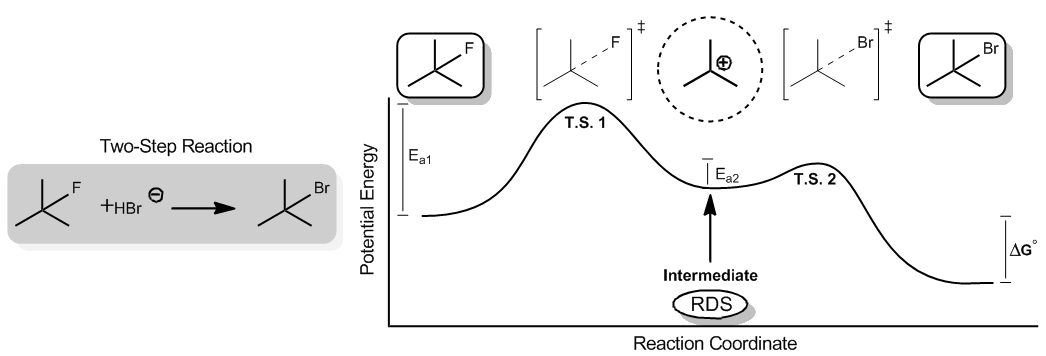
So it turns out that Delta G doesn't just doesn't just describe a one step reaction, it can also describe multi step reactions. All right, if you have more than one step to go to completion, the Delta G would just be the sum of all the steps. All right, Now, what I want to do is I want to show you guys a really common example of a two step reaction and show you what kind of species that generates. Okay, it turns out that here I have my two step reaction, um, shown you guys don't know this direction yet, so you don't need to actually understand it. But here is basically the reaction, the free energy diagram and the reaction Coordinate. What I start off with here is an alcohol Florida. Okay, then what I get is this transition state. Okay, see how says T s transition state, where the flooring is being partially broken right now. Okay, So what that means is it's this, like middle place, where the flooring is, like, not fully dissociated. But it's not also not fully associate ID, so it's like in the middle. Okay, then what I get is the species where the flooring is completely gone. And now I'm missing the octet of this carbon. This is called a Carvel cat on this happens to be what we call an intermediate. Okay, Then we get is another transition state where now we're going to try to put a Brahmin on here, right, Because the end product is that you have an alcohol bromine. So now, my bro main comes here, and it's partially making a bond. Okay, that's another transition state. And then finally, we have the last product, which is my alcohol promo. Okay, What I want to show you guys is the way that this actually relates to train what transition states are and what intermediates are. Okay. Transition states are high, very energetic points of the reaction that cannot be isolated. Okay, what does that mean In terms of can't be isolated, it means that if I have a test tube and I tried to get a bunch of this thing inside that test tube, it just wouldn't happen. And the reason is because it's a very, very high energy state. It's almost like if I was jumping into the air and falling back down the transition state is me being up in the air. Does that actually mean that I could ever just exist in the air for any given period of time? Not really. That's just a period of time that I have to go through in order to get to my end destination. But it doesn't. It's not actually something that you could isolate and just say, Okay, I want to see just Johnny up in the air for, like, five minutes. That's not gonna happen. Okay, um, than an intermediate is actually something that is a, ah, higher energy. But it can be isolated. Okay, It's a It's a molecule that is simply at a higher energy state than normal, but it actually does. Can be can exist, or it does can be isolated. So, for example, that would be like me jumping onto a stool. Okay, Now I have greater potential energy. I'm a little bit I don't have I'm not quite a stable because I have more potential energy. But I can stay there as long as I want. Okay. And that would be like this carbon Catalan right here. This car will. Catalan is not very. It's not as stable as the beginning. As you can see, the energy for the carb Acadian is greater than the energy for my starting re agent, which was the alcohol fluoride. But it's still stable enough that it could be isolated, and it could just exist there for any given period of time until I want to do something to it. Okay, so I just want to show you guys the difference between a transition state and an intermediate ah, transition state is always gonna be indicated by this little dagger sign at the top with brackets. Whenever you see that, that means that this is a transition state. Okay, An intermediate is usually not gonna have any kind of notation like that at all. It would just look like that. It will just the dotted line around. It has nothing to do with it. I'm just saying it might be like a positive charge. Whatever. Okay, Now, how are you going to notice the's on a free energy diagram? Because on a free energy energy diagram, these words that I gave you are these letters are not always gonna be given to you. Just know that the transition states are the highest energy states possible. So those are gonna be your highest points on the graph. Okay, So your transition states are always going to relate to the very highest energy points, the ones that you have to just pass through very quickly in order to get to the end. Okay, An intermediate is going to refer to any dip on the graph. Okay. Anytime they have a dip, that it's between two higher point. That's gonna be an intermediate. Okay, Now, notice that. Remember that we used to measure were we talked about how the rate of reaction had to do with the activation energy. But in this case, I actually have two different activation energies. I have the activation energy one that represents the amount of energy it takes to get first past my first transition state. Okay, but then I also have a second trend, a second activation energy here, which represents the amount of energy it takes to get over my second transition state. Okay, so which of these is going to tell me what the rate of the reaction is? Are they both do? I just add them up? And then that's the rate trick question absolutely not. Okay, What we're gonna do is we're just going to go with the highest activation energy. So what? Everyone, that is, whether it's activation energy. 123 or 10. Let's say it's a 10 step whatever. You just go with the very highest activation energy, and that is gonna be your rate determining step okay? Or what we call our slow step. So this would be my slow step right here. And what that means is that my rate determining step is gonna be forming this intermediate right there. Why is that? Because might notice that this activation energy is that high. This activation energy is tiny in comparison. So which one's gonna have a greater effect on the overall rate of the reaction? This one right here. Okay. And it turns out that in order to get to the end, the most determining thing is just gonna be how quickly can I go through the slow step? If I could make the slow step a little bit faster, that's good for my reaction in terms of rate. But it doesn't matter what I do to this activation energy. It's so small that it doesn't matter by comparison. I'm always gonna be waiting for the first one to form before I do the second one anyway. Okay. So I hope that that makes sense to you guys. I just want to show you guys the difference in a transition state and an intermediate. Once we get into actual reactions, this is gonna matter a whole lot because we have to talk about that all the time. In terms of what? The transition state and what's an intermediate? Alright. So hopeful. That makes sense. Let's go into the next topic.
Some reactions require more than one step to go to completion. The ΔGo is the sum of all the steps
Transition states are high-energy species that CANNOT be isolated. They involve bonds being broken and made at the same time.
Intermediates are high-energy species that CAN be isolated. They rest at a higher energy state than normal.
Do you want more practice?
We have more practice problems on Gibbs Free Energy